Current high throughput single cell and spatial transcriptomics use Illumina short-read to sequence cDNA templates in a very limited ( 50-100b) 3'part of the transcripts. Those methods have largely demonstrated a huge ability to decipher biological mechanisms at low resolution but the short-read sequencing behaviour misses important information than must be accessible through long-read sequencing of unfragmented same library preparation. Several companies have been developing long reads sequencers: instruments from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) can generate reads with a median length of 8-10 kb and as long as 100 kb, with a read length record of 4 Mb, obtained in September 2020 with a Nanopore sequencing system.
Due to the high throughput required for sequencing molecules from single cell experiment we choose Oxforde Nanopore Technology to generate long-reads. Nanopore technology identifies DNA bases by measuring the changes in electrical conductivity generated as DNA strands pass through a biological pore fixed on membrane in flow cells. When DNA molecules pass through or near the nanopore, there is a change in pattern or magnitude of the current in the nanopore, which can be observed, characterized and measured several thousand times per second by a sensor. The data streams are passed to the application-specific integrated circuit microchip (ASIC) and a software generates the signal-level data. We are now able to generate > 120 million cDNA long-read from a promethION ONT flow cell (feb.2021).
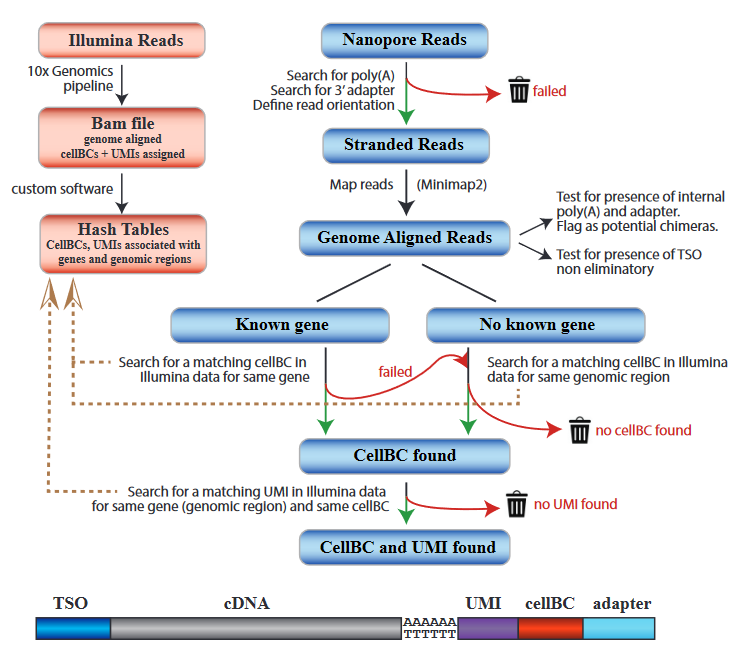
To ensure highly accurate barcode assignment, we used a strategy where barcodes assignment is guided with Illumina data.
We first extracted for each gene and genomic region (500 nt. windows) the barcodes detected in the Illumina short-read data.
We then compared the cell / spatial barcode sequence extracted from each genome aligned Nanopore read with the barcodes found in the Illumina data for the same gene or genomic region.
Following this strategy, we assigned cell / spatial barcode to 68% of Nanopore reads with identified poly(A) and adapter sequence. The poly(A) and cell / spatial barcode discovery rates
of our approach are, despite the higher error rates of Nanopore reads, similar to those reported previously for PacBio sequencing of 10x Genomics libraries.
After assignment of the cell / spatial barcode to the Nanopore read, we compared the Nanopore UMI read sequence with the UMI sequences found for the same gene (or genomic region) and the same cell
in the Illumina sequencing data. This strategy drastically reduces the complexity of the UMI search set, which corresponds to the number of transcripts molecules captured for a given gene or genomic
region in one cell. Using this strategy, we assigned UMIs to 76% of the reads with identified cell / spatial barcode.
Github v1 and v2 code repository: SiCeLoRe-2.0
SiCeLoRe repository release 2.1 now with short-read free analysis compatible with 10x Genomics Visium and single-cell 3' and 5' protocols.
Github v2.1 code repository: SiCeLoRe-2.1